Cowley short range order parameter
The Cowley short range order parameter can be used to find if an alloy is ordered or not. The order parameter is given by,
where \(n_i\) is the number of atoms of the non reference type among the \(c_i\) atoms in the \(i\)th shell. \(m_A\) is the concentration of the non reference atom.
We can start by importing the necessary modules
[1]:
import pyscal as pc
import pyscal.crystal_structures as pcs
import matplotlib.pyplot as plt
We need a binary alloy structure to calculate the order parameter. We will use the crystal structures modules to do this. Here, we will create a L12 structure.
[2]:
atoms, box = pcs.make_crystal('l12', lattice_constant=4.00, repetitions=[2,2,2])
In order to use the order parameter, we need to have two shells of neighbors around the atom. In order to get two shells of neighbors, we will first estimate a cutoff using the radial distribution function.
[4]:
sys = pc.System()
sys.box = box
sys.atoms = atoms
[5]:
val, dist = sys.calculate_rdf()
We can plot the rdf,
[7]:
plt.plot(dist, val)
plt.xlabel(r"distance $\AA$")
plt.ylabel(r"$g(r)$")
plt.xlim(0, 5)
[7]:
(0.0, 5.0)
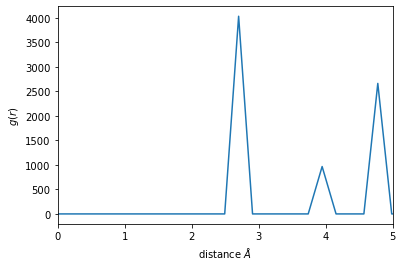
In this case, a cutoff of about 4.5 will make sure that two shells are included. Now the neighbors are calculated using this cutoff.
[8]:
sys.find_neighbors(method='cutoff', cutoff=4.5)
Finally we can calculate the short range order. We will use the reference type as 1 and also specify the average keyword as True. This will allow us to get an average value for the whole simulation box.
[9]:
sys.calculate_sro(reference_type=1, average=True)
[9]:
array([-0.33333333, 1. ])
Value for individual atoms can be accessed by,
[10]:
atoms = sys.atoms
[11]:
atoms[4].sro
[11]:
[-0.33333333333333326, 1.0]
Only atoms of the non reference type will have this value!