Analyzing a lammps trajectory
In this example, a lammps trajectory in dump-text format will be read in, and Steinhardt’s parameters will be calculated.
[1]:
import pyscal as pc
import os
import pyscal.traj_process as ptp
import matplotlib.pyplot as plt
import numpy as np
First, we will use the split_trajectory method from pyscal.traj_process module to help split the trajectory into individual snapshots.
[2]:
trajfile = "traj.light"
files = ptp.split_trajectory(trajfile)
files contain the individual time slices from the trajectory.
[3]:
len(files)
[3]:
10
[4]:
files[0]
[4]:
'traj.light.snap.0.dat'
Now we can make a small function which reads a single configuration and calculates \(q_6\) values.
[5]:
def calculate_q6(file, format="lammps-dump"):
sys = pc.System()
sys.read_inputfile(file, format=format)
sys.find_neighbors(method="cutoff", cutoff=0)
sys.calculate_q(6)
q6 = sys.get_qvals(6)
return q6
There are a couple of things of interest in the above function. The find_neighbors method finds the neighbors of the individual atoms. Here, an adaptive method is used, but, one can also use a fixed cutoff or Voronoi tessellation. Also only the unaveraged \(q_6\) values are calculated above. The averaged ones can be calculate using the averaged=True keyword in both calculate_q and get_qvals method. Now we can simply call the function for each file..
[6]:
q6s = [calculate_q6(file) for file in files]
We can now visualise the calculated values
[7]:
plt.plot(np.hstack(q6s))
[7]:
[<matplotlib.lines.Line2D at 0x7f5e424c0f60>]
Adding a clustering condition
We will now modify the above function to also find clusters which satisfy particular \(q_6\) value. But first, for a single file.
[8]:
sys = pc.System()
sys.read_inputfile(files[0])
sys.find_neighbors(method="cutoff", cutoff=0)
sys.calculate_q(6)
Now a clustering algorithm can be applied on top using the cluster_atoms method. cluster_atoms uses a condition as argument which should give a True/False value for each atom. Lets define a condition.
[9]:
def condition(atom):
return atom.get_q(6) > 0.5
The above function returns True for any atom which has a \(q_6\) value greater than 0.5 and False otherwise. Now we can call the cluster_atoms method.
[10]:
sys.cluster_atoms(condition)
[10]:
16
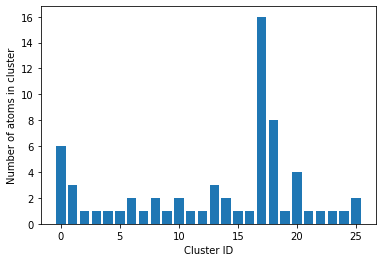
The method returns 16, which here is the size of the largest cluster of atoms which have \(q_6\) value of 0.5 or higher. If information about all clusters are required, that can also be accessed.
[11]:
atoms = sys.atoms
atom.cluster gives the number of the cluster that each atom belongs to. If the value is -1, the atom does not belong to any cluster, that is, the clustering condition was not met.
[12]:
clusters = [atom.cluster for atom in atoms if atom.cluster != -1]
Now we can see how many unique clusters are there, and what their sizes are.
[13]:
unique_clusters, counts = np.unique(clusters, return_counts=True)
counts contain all the necessary information. len(counts) will give the number of unique clusters.
[14]:
plt.bar(range(len(counts)), counts)
plt.ylabel("Number of atoms in cluster")
plt.xlabel("Cluster ID")
[14]:
Text(0.5, 0, 'Cluster ID')
Now we can finally put all of these together into a single function and run it over our individual time slices.
[15]:
def calculate_q6_cluster(file, cutoff_q6 = 0.5, format="lammps-dump"):
sys = pc.System()
sys.read_inputfile(file, format=format)
sys.find_neighbors(method="cutoff", cutoff=0)
sys.calculate_q(6)
def _condition(atom):
return atom.get_q(6) > cutoff_q6
sys.cluster_atoms(condition)
atoms = sys.atoms
clusters = [atom.cluster for atom in atoms if atom.cluster != -1]
unique_clusters, counts = np.unique(clusters, return_counts=True)
return counts
[16]:
q6clusters = [calculate_q6_cluster(file) for file in files]
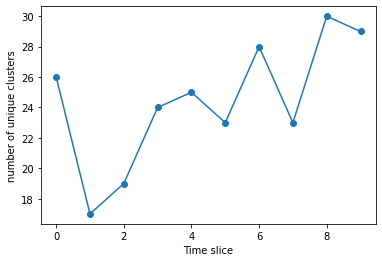
We can plot the number of clusters for each slice
[17]:
plt.plot(range(len(q6clusters)), [len(x) for x in q6clusters], 'o-')
plt.xlabel("Time slice")
plt.ylabel("number of unique clusters")
[17]:
Text(0, 0.5, 'number of unique clusters')
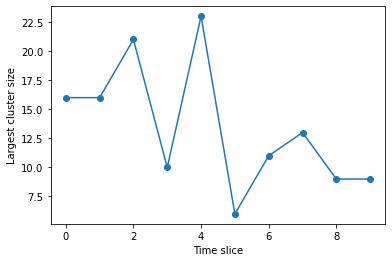
We can also plot the biggest cluster size
[18]:
plt.plot(range(len(q6clusters)), [max(x) for x in q6clusters], 'o-')
plt.xlabel("Time slice")
plt.ylabel("Largest cluster size")
[18]:
Text(0, 0.5, 'Largest cluster size')
The final thing to do is to remove the split files after use.
[19]:
for file in files:
os.remove(file)
Using ASE
The above example can also done using ASE. The ASE read method needs to be imported.
[20]:
from ase.io import read
[21]:
traj = read("traj.light", format="lammps-dump-text", index=":")
In the above function, index=":" tells ase to read the complete trajectory. The individual slices can now be accessed by indexing.
[22]:
traj[0]
[22]:
Atoms(symbols='H500', pbc=True, cell=[18.21922, 18.22509, 18.36899], momenta=...)
We can use the same functions as above, but by specifying a different file format.
[23]:
q6clusters_ase = [calculate_q6_cluster(x, format="ase") for x in traj]
We will plot and compare with the results from before,
[24]:
plt.plot(range(len(q6clusters_ase)), [max(x) for x in q6clusters_ase], 'o-')
plt.xlabel("Time slice")
plt.ylabel("Largest cluster size")
[24]:
Text(0, 0.5, 'Largest cluster size')
As expected, the results are identical for both calculations!